

Aspetti clinici, genetici e terapeutici della sindrome di Gorlin Goltz: revisione della letteratura e descrizione di un caso clinico rappresentativo
Paolo Ordesi1
Roberto Borloni2
Giovanni Felisati3
Paolo Lozza3
Maurizia Macchi2
Marco Persia2
Matteo Chiapasco4
Sandro Siervo2
1Allievo, Programma di formazione Postuniversitaria, Istituto Stomatologico Italiano, Milano
2Unità operativa di Chirurgia maxillo facciale, Direttore Dr. M. Macchi, Istituto Stomatologico Italiano, Milano
3Unità di Otorinolaringoiatria, Dipartimento di Medicina e Chirurgia, AO San Paolo, Università degli Studi di Milano
4Unità di Chirurgia Orale (direttore Professor Matteo Chiapasco) Dipartimento di Medicina, Chirurgia e Odontoiatria, AO San Paolo, Clinica Odontoiatrica, Università degli Studi di Milano
Riassunto
La sindrome di Gorlin Goltz, o Sindrome dei Nevi Basocellulari multipli, è un disordine caratterizzato dalla presenza di carcinomi basocellulari multipli, cheratocisti delle ossa mascellari, calcificazione della falce cerebrale, interstizi palmari e/o plantari. Unitamente a queste caratteristiche maggiori, sono stati descritti numerosi altri aspetti correlati alla sindrome quali costole bifide, labiopalatoschisi. La Sindrome è a trasmissione autosomica dominante, ad alta penetranza e variabile espressività, e si pone in essere per un alterazione a livello del gene PTCH del cromosoma 9 (9q22.3). Presso il Reparto di Chirurgia Maxillo-Facciale dell’Istituto Stomatologico Italiano di Milano è stato reclutato un paziente che presentava un quadro, poi confermato, di Sindrome di Gorlin-Goltz. Il paziente è stato sottoposto a un totale di 3 interventi per l’enucleazione di 4 cheratocisti dei mascellari, di cui una recidivante, e di 3 interventi per l’asportazione di 4 carcinomi a cellule basali a livello del volto. L’analisi genetica non ha dimostrato apparenti alterazioni a livello del gene PTCH. La diagnosi di Sindrome di Gorlin Goltz è unicamente clinica, e prevede dei criteri d’inclusione maggiori e minori.
La gestione terapeutica delle lesioni a più alto tasso di recidiva caratteristiche della sindrome, come i carcinomi a cellule basali e le cheratocisti odontogene, prevede la loro enucleazione chirurgica. In entrambi i casi, la letteratura ha descritto differenti e validi approcci per la loro asportazione.
Parole chiave: Sindrome di Gorlin-Goltz, cheratocisti odontogena, carcinoma a cellule basali
Summary
Clinical, genetic and therapeutic aspects of the Gorlin-Goltz Syndrome: review of the literature and a case report
Gorlin-Goltz syndrome, Nevoid basal cell carcinoma syndrome (NBCCS), is a disorder characterized by the presence of multiple basal cell carcinomas, keratocysts of the jaws, calcification of the falx cerebri, palmar and plantar pits. Together with these major features have been described many other aspects related to the syndrome, such as bifid ribs, cleft lip and palate. The syndrome is autosomal dominant, with high penetrance and variable expressivity, and is related to alterations of the PTCH gene on chromosome 9 (9q22.3). At the department of Maxillofacial Surgery of the Istituto Stomatologico Italiano of Milano was recruited a patient who presented a clinical situation, later confirmed, of Gorlin-Goltz syndrome. The patient was subjected to a total of 3 interventions for enucleation of 4 keratocysts of the jaws, one of which recurred, and 3 interventions for the removal of 4 basal cell carcinomas at the level of the face. Genetic analysis did not demonstrate apparent alterations in the PTCH gene. The diagnosis of Gorlin-Goltz syndrome is only clinical, including major and minor criteria.
The therapeutic management of the high rate of recurrence lesions of the syndrome, such as basal cell carcinomas and odontogenic keratocysts, requires their surgical enucleation. In both cases, the literature has described different and valid approaches for their removal.
Key words: Gorlin-Goltz Syndrome, odontogenic keratocyst, basal cell carcinoma
La sindrome di Gorlin Goltz, denominata anche sindrome dei carcinomi basocellulari nevoidi (NBCCS), è un disordine a trasmissione autosomica dominante ad alta penetranza ed espressività variabile, caratterizzato da una costellazione di segni clinici e dalla predisposizione allo sviluppo di determinate neoplasie1. Ha una prevalenza che la letteratura ha stimato da 1 individuo su 57.0002-4 sino a 1 individuo su 164.0005. Uno studio attesta la prevalenza italiana in 1 individuo su 256.0006. Clinicamente la sindrome è stata delineata da Gorlin e Goltz nel 1960, descrivendo la classica triade (segni maggiori), composta da carcinomi a cellule basali multipli a livello della testa e del collo, cheratocisti odontogene (KCOT) delle ossa mascellari e pits palmo plantari1. I carcinomi a cellule basali sono neoformazioni che possono svilupparsi sul tronco e sul collo. Possono variare in numero da poche unità a diverse migliaia, di dimensioni che vanno da 1 a 10 millimetri di diametro.
L’incidenza negli individui caucasici è maggiore rispetto a quella dei soggetti afro-americani, probabilmente per l’azione protettiva dai raggi UV che la melanina svolge in questi ultimi7-9. Le cheratocisti odontogene (KCOT) recidivanti rappresentano un segno clinico patognomonico della sindrome. Si riscontrano dal 66% al 90% dei casi. Sono il segno più consistente nei pazienti giovani: il 13% dei soggetti le sviluppa entro la prima decade di vita, e il 51% entro la seconda5,10. Il loro riscontro è generalmente casuale durante lo svolgimento di un’ortopantomografia o di una teleradiografia. Di regola asintomatiche, danno manifestazione di sé in caso di sovrainfezione, compressione nervosa, mobilità dentaria o sanguinamento11,12. Il sito d’insorgenza più frequente è il ramo mandibolare (44%), seguito dalla regione incisivo canina mandibolare (18%) e mascellare (15%) e, infine, dal tuber (13%)6. Grave problema correlato alle cheratocisti odontogene è il loro alto tasso di recidiva (60%), probabilmente dovuto alle loro peculiarità istologiche13,14.
La sindrome di Gorlin Goltz è caratterizzata da calcificazioni ectopiche del sistema nervoso centrale, quali la calcificazione lamellare della falce cerebrale, del tentorio cerebellare, della sella turcica, e del suo diaframma6,15,16,53. Di riscontro occasionale in quanto asintomatiche, sono di estremo ausilio per l’inquadramento diagnostico della sindrome. Altre situazioni patologiche riscontrabili (segni minori) sono macrocefalia, labiopalatoschisi, ipertelorismo, bozze frontali, anomalia di Sprengel, ovvero un’elevazione congenita della scapola dovuta a mancata discesa della stessa nella sua normale posizione durante la vita fetale, sindattilia, anomalie vertebrali, calcificazione del legamento interclinoideo, fibromi ovarici, medulloblastomi. La maggior parte delle lesioni correlate alla sindrome non è letale. Il medulloblastoma può essere l’unica causa di morte precoce, anche se le percentuali di sopravvivenza al tumore sono superiori nei soggetti sindromici rispetto a persone non affette dalla sindrome malate di medulloblastoma6.
Studi genetici hanno dimostrato una correlazione tra l’insorgenza della sindrome e un’alterazione a livello del gene PTCH (9q22.3)17-24. Il gene PTCH codifica una proteina transmembrana, con funzione di oncosoppressore, che fa parte del Sistema di Segnalazione Hedgehog, un sistema a secondo messaggero che svolge un ruolo chiave nell’embriogenesi dei vertebrati25-28. In particolare, quando la proteina PTCH si lega con il messaggero SHH (Sonic Hedgehog), interrompe la propria azione inibitoria che ha su un’altra proteina transmembrana, SMO (Smoothened), la quale attiva i fattori nucleari di trascrizione gli. Il ruolo morfogenetico di questa via è stato avvalorato da studi che hanno dimostrato come l’identità e lo sviluppo degli elementi dell’arto siano in funzione della concentrazione e del tempo di esposizione di SHH52.
Descrizione del caso clinico
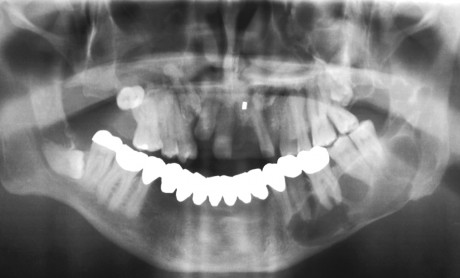
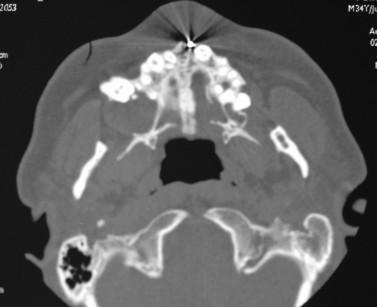
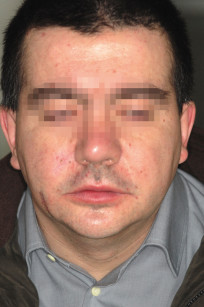
Il caso presentato in questo articolo si riferisce a un paziente maschio, di 37 anni di età al momento del reclutamento, di etnia caucasica. È stato inviato presso la nostra unità operativa di chirurgia maxillo-facciale nel mese di novembre 2007. Al momento della visita il paziente riportava una costellazione di sintomi correlabili alla sindrome. In particolare, era riscontrabile radiologicamente la presenza di 3 ampie neoformazioni di natura cistica, a livello del mascellare superiore bilateralmente, in regione 16-18 e 23-27, e a livello dell’emimandibola sinistra, a partire dall’elemento 34 fino alla branca montante omolaterale. In senso cranio-caudale, la neoformazione occupava la quasi totalità del corpo mandibolare, anche se l’esame TC escludeva l’erosione di entrambe le corticali ossee. La neoformazione sita a livello del mascellare superiore destro aveva provocato l’erosione del corpo mascellare, sconfinando e occupando interamente il seno mascellare omolaterale. Era inoltre evidenziabile un grado di erosione della parete laterale della cavità nasale destra. Il paziente riportava in anamnesi numerosi interventi di asportazione di carcinomi a cellule basali a livello del capo e del collo.
Al momento della visita, era presente una neoformazione a livello della guancia destra. A livello clinico, il paziente inoltre presentava una calcificazione della falx cerebri, riscontrata con l’esecuzione della TC del massiccio facciale, e un ispessimento della diploe. Erano presenti una facies caratteristica, con bozze frontali associate a fronte alta e larga, ipertelorismo, radice del naso allargata, pits palmari. A livello anamnestico, il paziente dichiarava di essere stato sottoposto a 3 interventi di enucleazione di neoformazioni cistiche delle ossa mascellari nel 1981, 1983 e 1985. Negava lo sviluppo di medulloblastoma in età giovanile e un riscontro all’interno della famiglia della medesima situazione clinica. Nel gennaio del 2008 il paziente è stato sottoposto a intervento di asportazione della neoformazione cistica a livello dell’osso mascellare superiore destro. È stato effettuato un trattamento chirurgico combinato, con accesso sia vestibolare che nasale con tecnica chirurgica FESS (functional endoscopic sinus surgery), per la rimozione della componente nasale della neoformazione. Contestualmente, sono stati estratti gli elementi dentari 17, 16 e 13, a stretto contatto con la stessa. L’approccio è stato di tipo conservativo, con accurata toilette dei margini ossei, senza resezione en-bloc preventiva. L’esame istologico ha confermato la diagnosi di cheratocisti odontogena.
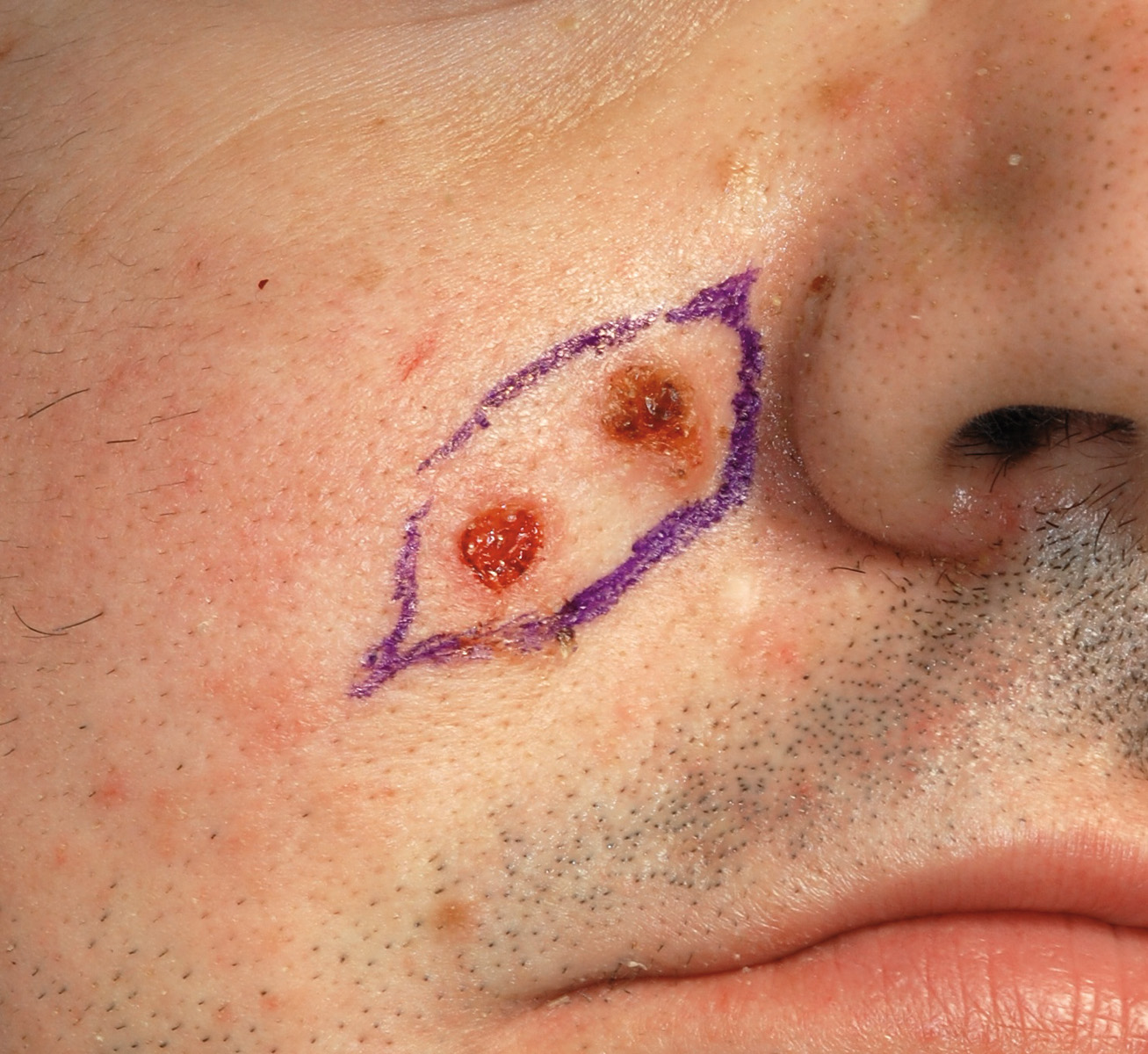
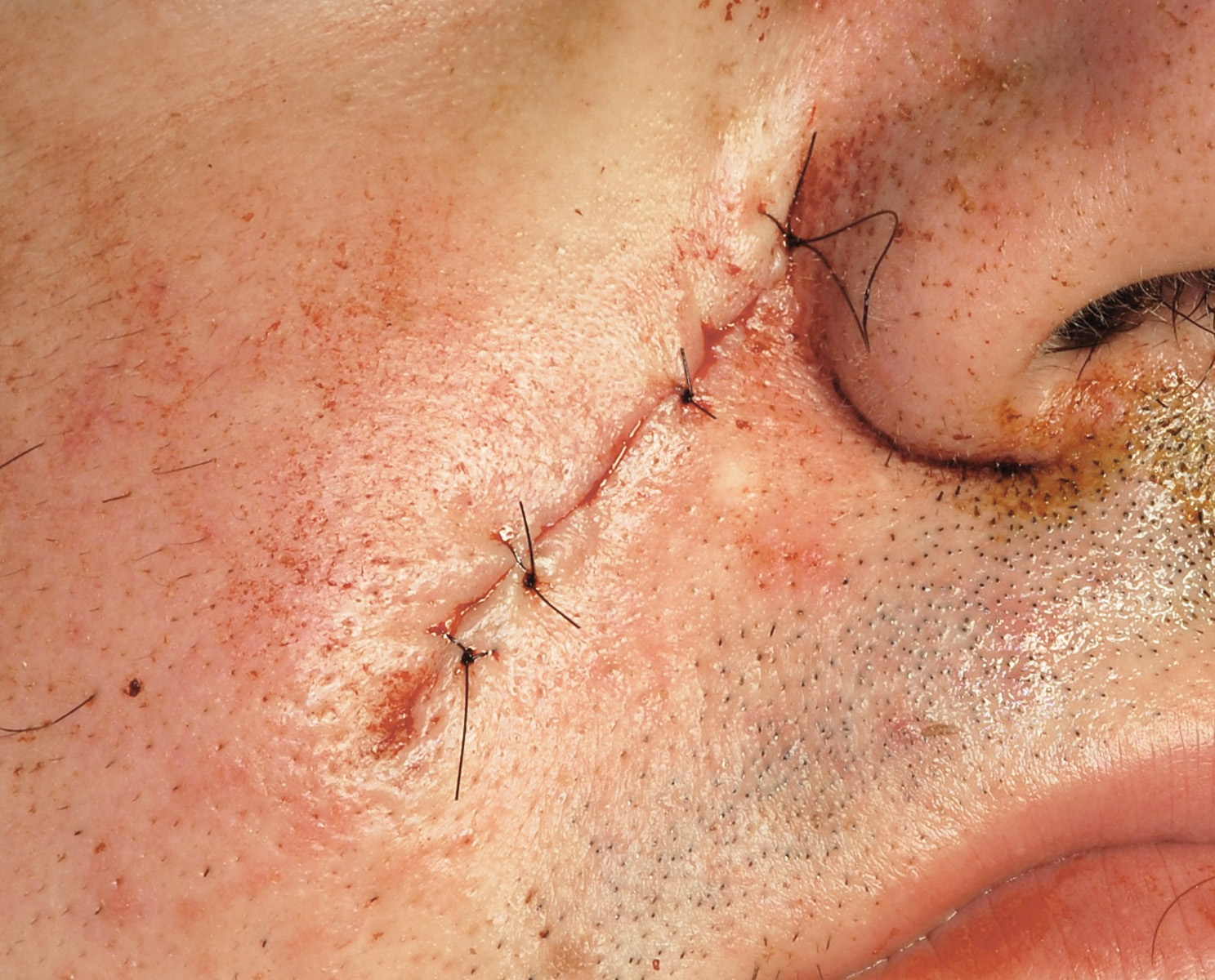
Nell’aprile 2008 il paziente è stato sottoposto a intervento di asportazione di carcinoma a cellule basali a livello del solco nasogenieno destro. Nel luglio dello stesso anno il paziente è stato sottoposto a intervento di asportazione delle neoformazioni cistiche mascellare superiore sinistra e mandibolare. Anche in questo caso, l’approccio chirurgico è stato di tipo conservativo. Nel novembre 2008 il paziente è stato sottoposto a intervento di asportazione di un singolo carcinoma a cellule basali a livello della guancia destra, a 1 cm dalla rima labiale omolaterale, comparso nel maggio del 2008. Nel giugno del 2009 il paziente è stato sottoposto a intervento di asportazione della neoformazione mascellare superiore sinistra recidivata. Allestito un lembo di tipo trapezoidale, ed effettuata la scheletrizzazione, è stata asportata attraverso l’accesso osseo creato nel precedente intervento la neoformazione recidivante. Contestualmente, sono stati estratti gli elementi 26 e 27 le cui radici risultavano a contatto con la neoformazione stessa.
Nell’ottobre 2009 il paziente è stato sottoposto a intervento di asportazione di due carcinomi a cellule basali rispettivamente in regione fronto-temporale e malare destra. Nel giugno 2009 è stata effettuata l’analisi della sequenza del gene PTCH. Prelevato un campione di 8 ml di sangue intero, dalla linea linfoblastoide è stato estratto il Dna (NUCLEON BACC3), che successivamente è stato amplificato tramite PCR. La miscela è stata sottoposta a una serie di cicli di attivazione a temperature stabilite (9’ di attivazione a 95 °C, seguiti da 40 cicli di 50” a 95 °C, 30” a 55-60 °C, 1’ a 72 °C, 10’ a 72 °C per l’estensione finale). Il risultato, purificato, è stato sottoposto a sequenziazione genica. Dal punto di vista genetico, il paziente non ha mostrato alcuna alterazione della sequenza codificante del gene. Anche l’analisi dei promotori alternativi non ha evidenziato alcuna mutazione. L’esame all’MLPA delle microdelezioni intrageniche ha riscontrato dei Polimorfismi a Singolo Nucleotide (SNP) in eterozigosi (Tabella 3-1)29-32.
Discussione e conclusioni
La diagnosi di sindrome di Gorlin Goltz è prettamente clinica ed è possibile identificare un caso in presenza di 2 criteri maggiori, oppure di 1 criterio maggiore e 2 minori9: vediamoli. Criteri maggiori:
- carcinomi basocellulari multipli (>2 o almeno 1 in pazienti al di sotto dei 20 anni di età);
- cheratocisti odontogene delle ossa mascellari diagnosticate istologicamente;
- pits palmoplantari (almeno 3);
- calcificazione bilamellare della falce cerebrale;
- coste bifide o fuse;
- parenti di primo grado affetti da sindrome di Gorlin Goltz.
Criteri minori:
- macrocefalia (determinata al termine dello sviluppo);
- malformazioni congenite, come labiopalatoschisi, ipertelorismo, bozze frontali, “faccia grossa”;
- altre anomalie scheletriche, come deformità di Sprengel, marcate deformità del petto, sindattilia;
- anomalie radiologiche: “sella turcica bridging” (calcificazione del legamento interclinoideo), anomalie vertebrali come emivertebre, fusione o allungamento dei corpi vertebrali, difetti di forma di mani e piedi, trasparenze a forma di fiamma di mani e piedi;
- fibroma ovarico;
- medulloblastoma.
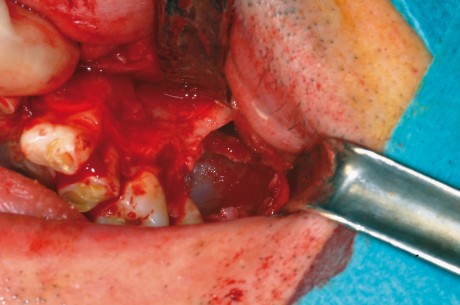
Al momento della prima visita il nostro paziente presentava 4 criteri maggiori (cheratocisti odontogene, carcinomi a cellule basali, calcificazione della falce cerebrale, pits palmari) e 5 criteri minori (ispessimento della diploe, ipertelorismo, facies caratteristica, bozze frontali associate a fronte alta e larga, radice del naso allargata). L’asportazione chirurgica dei carcinomi a cellule basali è la terapia da privilegiarsi in un paziente caratterizzato da un quadro di carcinomi a cellule basali limitato in numero. Questo tipo di chirurgia permette un’asportazione radicale della lesione, con un rischio minimo di complicanze, un’elevata percentuale di successo e un ottimo risultato estetico. L’asportazione a lama fredda consente un preciso controllo istologico dei margini resecati. La letteratura cita come complicanze, seppur rare, la formazione di cheloidi o lesioni a strutture nervose53.
A latere della enucleazione a lama fredda, la letteratura ha descritto numerose altre terapie efficaci nell’enucleazione dei carcinomi, come la terapia fotodinamica33,35-37,53, la chemioterapia topica, l’ablazione a mezzo di laser, la criochirurgia53 e l’elettrodessicazione 34-53. L’utilizzo di queste terapie è stato indicato generalmente per la cura di lesioni numerose, di piccole dimensioni, localizzate in aree a bassa valenza estetica e a basso indice di ricorrenza come il tronco. Nel nostro caso, il paziente ha sviluppato, durante il nostro periodo d’osservazione, un totale di 3 lesioni, di medie dimensioni, site sul volto. Pertanto, per il numero, le dimensioni e la localizzazione dei carcinomi a cellule basali, l’approccio a lama fredda è stato l’unico preso in considerazione. Per quanto riguarda la gestione non chirurgica dei carcinomi a cellule basali, sono state descritte sia terapie squisitamente farmacologiche, mediante l’utilizzo di retinoidi o interferone, che in supporto alla terapia chirurgica, con l’utilizzo di etretinato. In entrambi i casi, la letteratura ha mostrato dei risultati non univoci38-41. La gestione chirurgica delle cheratocisti odontogene è un argomento ancora fonte di dibattito in letteratura. Esistono due tipi di approccio: il primo, conservativo, consiste nella semplice rimozione della lesione, eventualmente preceduta da marsupializzazione.
Il secondo, aggressivo, prevede la resezione dei margini ossei a contatto con la neoformazione e l’utilizzo della soluzione di Carnoy42. In molti casi l’approccio conservativo viene giudicato come il più indicato, considerando il danno biologico arrecato da un intervento radicale43,44. Numerosi Autori mostrano come la tecnica di marsupializzazione offra un’alta percentuale di successo, con un tasso di recidiva inferiore rispetto all’approccio puramente conservativo soprattutto in casi di cheratocisti multiple45,46. Per il ridotto danno biologico, l’utilizzo della marsupializzazione è indicato soprattutto nei pazienti pediatrici44,47. Un approccio chirurgico aggressivo, caratterizzato dalla resezione en-bloc dei margini ossei coinvolti dovrebbe essere tenuto in considerazione nei seguenti casi: recidiva di una cheratocisti in caso di sua precedente enucleazione con procedure alternative; recidiva in caso di marsupializzazione; presenza di cheratocisti multiloculari; lesioni dall’aspetto clinico particolarmente aggressivo43.
I risultati genetici da noi ottenuti non sono in grado di riscontrare una ben precisa correlazione della malattia. La letteratura ha mostrato come alla base della sindrome sia presente nell’85% dei casi una mutazione del gene PTCH, e come la mutazione più frequentemente riscontrata sia una troncazione48-50. Il soggetto rientra nel 15% dei pazienti apparentemente indenni da alterazioni a carico del gene PTCH. Questi dati dimostrano come in realtà i meccanismi che regolano l’Hedgehog Signaling Network e le alterazioni a esso correlate siano ancora poco conosciuti. A dimostrazione di ciò, gli studi sinora documentati non hanno riscontrato delle correlazioni precise tra alterazione genotipica e manifestazione fenotipica48. Pertanto, la patogenesi della sindrome è ipotizzabile che sia correlata a un’alterazione di componenti della rete posti a valle rispetto a PTCH, quali le proteine GLI (GLI1-2-3) e SUFU51. La gestione dei pazienti affetti da sindrome di Gorlin Goltz comporta un programma di visite dermatologiche regolari (ogni 2-3 mesi), specialmente durante la pubertà e l’adolescenza36. Concludendo, il nostro caso dimostra come la sindrome di Gorlin Goltz sia un quadro patologico complesso, che necessita di un corretto inquadramento diagnostico e di una collaborazione interdisciplinare per la gestione di tutti gli aspetti patologici che la caratterizzano.
Corrispondenza
paoloroberto.ordesi@gmail.com
bibliografia
1. Gorlin R.J, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med 1960;262:908-12.
2. Pratt MD, Jackson R. Nevoid basal cell carcinoma syndrome. A 15-year follow-up of cases in Ottawa and the Ottawa Valley. J Am Acad Dermatol 1987;16(5 Pt 1):964-70.
3. Evans DG, Birch JM, et al.Brain tumours and the occurrence of severe invasive basal cell carcinoma in first degree relatives with Gorlin syndrome. Br J Neurosurg 1991;5(6):643-6.
4. Farndon PA, Del Mastro RG, et al. Location of gene for Gorlin syndrome. Lancet 1992;339(8793):581-2.
5. Shanley S, Ratcliffe J, et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 1994;50(3):282-90.
6. Lo Muzio L, Nocini PF, et al. Nevoid basal cell carcinoma syndrome. Clinical findings in 37 Italian affected individuals. Clin Genet 1999;55(1):34-40.
7. Lo Muzio L, Nocini P, et al. Early diagnosis of nevoid basal cell carcinoma syndrome. J Am Dent Assoc 1999;130(5):669-74.
8. Goldstein AM, Pastakia B, et al. Clinical findings in two African-American families with the nevoid basal cell carcinoma syndrome (NBCC). Am J Med Genet 1994;50(3):272-81.
9. Kimonis VE, Goldstein AM, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 1997;69(3):299-308.
10. Evans DG, Ladusans EJ, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 1993;30(6):460-4.
11. Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98-113.
12. Blanchard SB. Odontogenic keratocysts: review of the literature and report of a case. J Periodontol 1997:68:306-311.
13. Dominguez FV, Keszler A Comparative study of keratocysts, associated and non-associated with nevoid basal cell carcinoma syndrome. J Oral Pathol 1988;17(1):39-42.
14. Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med 2004;6(6):530-9.
15. Ratcliffe JF, Shanley S, et al. The diagnostic implication of falcine calcification on plain skull radiographs of patients with basal cell naevus syndrome and the incidence of falcine calcification in their relatives and two control groups. Br J Radiol 1995;68(808):361-8.
16. Takanashi J, Fujii K, et al. Empty sella syndrome in nevoid basal cell carcinoma syndrome. Brain Dev 2000;22(4):272-4.
17. Towns TM, Lagattuta V Basal cell nevus syndrome: 20-year follow-up. J Oral Surg 1974;32(1):50-3.
18. Hahn H, Wicking C, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996;85(6):841-51.
19. Johnson RL, ARothman AL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996;272(5268):1668-71.
20. Unden AB, Holmberg E, et al. Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res 1996;56(20):4562-5.
21. Unden AB, Stahle-Backdahl M, et al. Fine mapping of the locus for nevoid basal cell carcinoma syndrome on chromosome 9q. Acta Derm Venereol 1997;77(1):4-9.
22. Unden AB., Zaphiropoulos PG, et al. Human patched (PTCH) mRNA is overexpressed consistently in tumor cells of both familial and sporadic basal cell carcinoma. Cancer Res 1997;57(12):2336-40.
23. Wicking C, Shanley S, et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet 1997;60(1):21-6.
24. Minami M, Urano Y, et al. Germline mutations of the PTCH gene in Japanese patients with nevoid basal cell carcinoma syndrome. J Dermatol Sci 2001;27(1):21-6.
25. Ingham PW, McMahon AP Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001;15(23):3059-87.
26. Bhardwaj G, Murdoch B, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol 2001;2(2):172-80.
27. Ahn S, Joyner AL In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 2005;437(7060):894-7.
28. Paladini RD, Saleh J, et al. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol 2005;125(4):638-46.
29. Eklund LK, Lindstrom E, et al. Mutation analysis of the human homologue of Drosophila patched and the xeroderma pigmentosum complementation group A genes in squamous cell carcinomas of the skin. Mol Carcinog 1998;21(2):87-92.
30. Pastorino L, Cusano R, et al. Molecular characterization of Italian nevoid basal cell carcinoma syndrome patients. Hum Mutat 2005;25(3):322-3.
31. Reifenberger J, Wolter M, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol 2005;152(1):43-51.
32. Mansilla MA, Cooper ME, et al. Contributions of PTCH gene variants to isolated cleft lip and palate. Cleft Palate Craniofac J 2006;43(1):21-9.
33. Oseroff AR, Blumenson LR, et al. A dose ranging study of photodynamic therapy with porfimer sodium (Photofrin) for treatment of basal cell carcinoma. Lasers Surg Med 2006;38(5):417-26.
34. Southwick GJ, Schwartz RA. The basal cell nevus syndrome: disasters occurring among a series of 36 patients. Cancer 1979;44(6):2294-305.
35. Grobbelaar AO, Horlock N, et al. Gorlin’s syndrome: the role of the carbon dioxide laser in patient management. Ann Plast Surg 1997;39(4):366-73.
36. Nouri K, Chang A, et al. Ultrapulse CO2 used for the successful treatment of basal cell carcinomas found in patients with basal cell nevus syndrome. Dermatol Surg 2002;28(3):287-90.
37. Doctoroff A, Oberlender SA, et al. Full-face carbon dioxide laser resurfacing in the management of a patient with the nevoid basal cell carcinoma syndrome. Dermatol Surg 2003;29(12):1236-40.
38. Gorlin RJ. Nevoid basal cell carcinoma syndrome. Dermatol Clin 1995;13(1):113-25.
39. Sanchez-Conejo-Mir J, Camacho F. Nevoid basal cell carcinoma syndrome: combined etretinate and surgical treatment. J Dermatol Surg Oncol 1989;15(8):868-71.
40. Peck GL, DiGiovanna JJ, et al. Treatment and prevention of basal cell carcinoma with oral isotretinoin. J Am Acad Dermatol 1988;19(1 Pt 2):176-85.
41. Neville JA, Williford PM, et al. Pilot study using topical imiquimod 5% cream in the treatment of nodular basal cell carcinoma after initial treatment with curettage. J Drugs Dermatol 2007;6(9):910-4.
42. Casaroto AR, Loures DC, Moreschi E, Veltrini VC, Trento CL, Gottardo VD, Lara VS. Early diagnosis of Gorlin-Goltz syndrome: case report. Head Face Med 2011 Jan 25;7:2.
43. Tolstunov L, Treasure T. Surgical treatment algorithm for odontogenic keratocyst: combined treatment of odontogenic keratocyst and mandibular defect with marsupialization, enucleation, iliac crest bone graft, and dental implants. J Oral Maxillofac Surg 2008;66:1025-36.
44. Habibi A, Saghravanian N, Habibi M, Mellati E, Habibi M. Keratocystic odontogenic tumor: a 10-year retrospective study of 83 cases in an Iranian population. J Oral Sci 2007;49:229-35.
45. Maurette PE, Jorge J, De Moraes M. Conservative treatment protocol of odontogenic keratocyst: a preliminary study. J Oral Maxillofac Surg 2006;64:379-83.
46. Pogrel MA. Treatment of Keratocysts: the case for decompression and marsupialization. J Oral Maxillofac Surg 2005;63:667-73.
47. Nakamura N, Mitsuyasu T, Mitsuyasu Y, Taketomi T, Higuchi Y, Ohishi M. Marsupialization for odontogenic keratocysts: Long-term follow-up analysis of the effects and changes in growth characteristics. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002;94:543-53.
48. Wicking C, Shanley S, et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet 1997;60(1):21-6.
49. Johnson RL, Rothman AL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996;272(5268):1668-71.
50. Hahn H, Wicking C, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996;85(6):841-51.
51, Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garrè ML, Clementi M, Scarrà GB. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 2009 Jul;149A(7):1539-43.
52. Harfe BD, Scherz PJ, et al. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004;118(4):517-28.
53. Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J Rare Dis 2008 Nov 25;3:32. Review.



{kind=link}