


Francisco Guinot Jimeno1
Raquel Torrents Gras1
Cristina Cuadros Fernandez2
Ana Isabel Lorente Rodriguez3
1Profesor/a asociado/a del Departamento de Odontopediatría - Universitat Internacional de Catalunya, Facultad de Ciencias de la Salud
2Directora del Máster de Odontopediatría Integral - Universitat Internacional de Catalunya, Facultad de Ciencias de la Salud
3Jefa del Departamento de Odontopediatría - Universitat Internacional de Catalunya, Facultad de Ciencias de la Salud
La sindrome di Noonan (SN) era conosciuta in passato come sindrome Ullrich-Turner. Furono questi due ricercatori, Ullrich nel 1930 e Turner nel 1938, a individuare un gruppo di donne e uomini con una sindrome caratterizzata da infantilismo sessuale e anomalie minori, tra cui una delle più comuni era lo pterigio del collo o collo alato2,4.
Nel 1883, Kobyslinsky descrisse il caso di un uomo giovane con le caratteristiche essenziali di questa sindrome, ma bisogna attendere il 1963 per ottenere ufficialmente il riconoscimento del quadro sindromico pediatrico cardiologico grazie a Noonan ed Ehmke. Gli Autori descriveranno infatti una serie di pazienti affetti da multiple malformazioni, includendo difetti congeniti del cuore e rare fasciti muscolari2,4,5.
Eziologia
L’eziologia della sindrome di Noonan è tuttora poco chiara, considerato che si eredita in forma autosomica dominante, con predominio di trasmissione materna, anche se nel 60% dei casi appare anche in forma sporadica1,2. Nel 2001, Tartaglia et al.6 hanno scoperto una serie di mutazioni del gene PTPN11 presente nel cromosoma 12q24.1 nel 50% dei soggetti studiati3,7,8. Questo gene è il responsabile della produzione di una proteina chiamata SHP-2, fondamentale per la trasmissione di diversi segnali intracellulari e per il controllo dello sviluppo del mesodermico e della genesi della valvola cardiaca semilunare. Questa proteina si produce in tutto il corpo e ha un ruolo importante nella risposta cellulare correlata ai fattori di accrescimento. Le mutazioni di PTN11 presenti nella SN danno luogo a un aumento della proteina SHP-2 e a un’alterata regolazione delle vie di trasduzione dei segnali cellulari attivati dalla proteina Ras e dalle protein-chinasi attivate da mitogeni (MAPK)9. La via di segnalazione Ras-MAPK produce segnali per i fattori di accrescimento e citochine che partecipano nella trascrizione dei geni coinvolti nella proliferazione cellulare e nella determinazione della differenziazione cellulare.
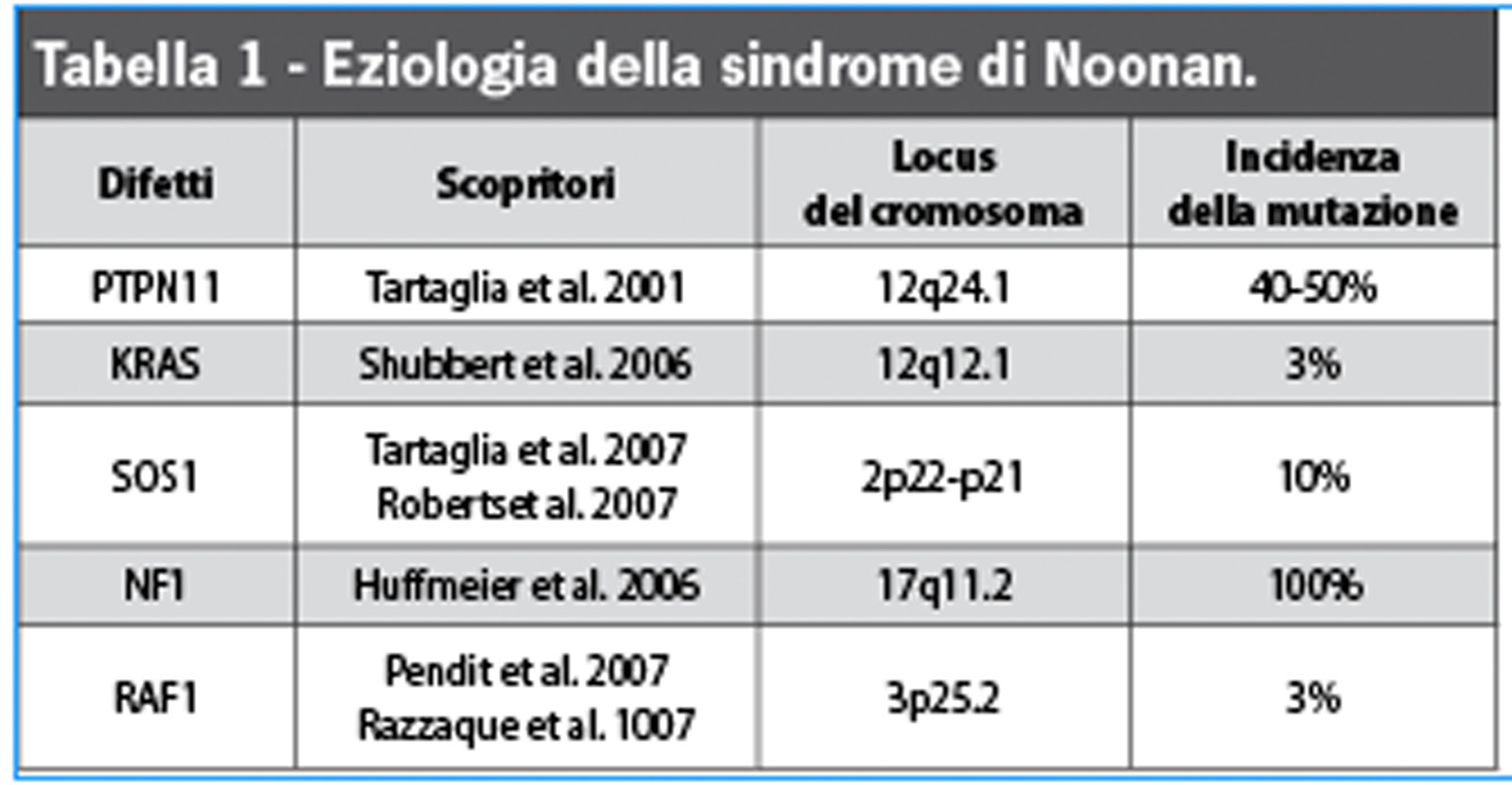
La via Ras-MAPK è più conosciuta nella patogenesi del cancro2,10-12. Nel 2005 Shubbert et al.13 scoprirono mutazioni in un secondo gene in pazienti affetti da SN. Questo gene è stato chiamato KRAS, ed è responsabile dell’aumento della funzione della proteina SHP-2 e della deregolazione della via Ras-MAPK. Le mutazioni di questo gene sono responsabili di meno del 5% dei casi di SN11. All’inizio del 2007 si scoprì la mutazione del gene SOS1, che si presenta nel 15% dei casi, codificante delle proteine implicate nella via Ras, causando la deregolazione della via e un aumento del segnale di trasduzione. I pazienti che presentano questa mutazione hanno un aumento dell’incidenza di sembianze ectodermiche poco comuni (cheratosi pilare facciale), capelli ricci e statura normale11. Esistono mutazioni in un quarto gene implicato (NF1), che causano la perdita della funzione della proteina neurofibromina dando luogo alla neurofibromatosi tipo 1, malattia associata a ritardo mentale, neurofibromi, macchie cutanee caffelatte, noduli fibrosi dell’iride e una tendenza a sviluppare tumori maligni11. Un quinto gene (RAF1) è stato identificato come causante della SN, ed è stato messo in relazione con il presentarsi della cardiomiopatia ipertrofica4,14,15 (Tabella 1).
Incidenza
L’incidenza della sindrome di Noonan è sconosciuta, si valuta che oscilli da 1:1000 a 1:2500 nati vivi2, anche se Jamieson et al.7 descrivono un’incidenza più bassa, intorno a 1:5000 nati vivi.
Caratteristiche cliniche
Nella Tabella 2 vengono descritte le caratteristiche cliniche più importanti della SN.
Il dismorfismo facciale tipico della sindrome di Noonan si modifica durante la crescita: nel neonato osserviamo un’eccessiva quantità di pelle sulla nuca, come risultato di un igroma cistico fetale. A 3 o 4 anni il corpo è più robusto e il seno più prominente. In questo periodo le deformazioni del torace incominciano a essere più significative.
Durante l’infanzia la testa è relativamente grande, prominente e rotonda e presenta occhi con ipertelorismo. Il ponte nasale solitamente è piatto, le orecchie sono lievemente postero-rotate e con inserzione bassa, il collo è generalmente corto con la persistenza di pterigio o collo alato. Altri segni facciali tipici di questa età sono: pieghe epicantiche, fessure palpebrali oblique, palato ogivale e micrognazia9,11.
Nell’adolescenza l’apparenza facciale incomincia a mostrare tratti più grossolani, il viso appare triangolare e il mento sembra più allungato.
Nell’età adulta il dimorfismo facciale si riduce; restano però un solco nasolabiale prominente e uno pterigio del collo, solitamente i capelli sono più radi e la loro linea di inserzione frontale è più alta, dando l’impressione di una fronte più ampia. I tratti facciali durante la vecchiaia possono essere più attenuati9,11.
Una delle caratteristiche cliniche più frequenti, associata al 70% dei casi, è la bassa statura. Alla nascita, il peso e la statura sono normali, anche se durante il primo anno di vita la crescita risulta ridotta. Viene segnalato un ritardo puberale di circa due anni. Nell’età adulta, la statura solitamente è di circa 160 cm per gli uomini e 150 cm per il sesso femminile1,11, ma restano sconosciuti i fattori causali: una possibile ipotesi sarebbe la secrezione insufficiente dell’ormone della crescita (GH) o del suo realising factor11,16; esistono però pareri controversi sugli effetti della terapia con GH in relazione con l’aumento della statura in età adulta4,9,17,18.
Altro segno clinico presente nell’80% dei pazienti affetti da SN sono i difetti congeniti del cuore, essendo la stenosi della valvola polmonare (50-62%) e la cardiomiopatia ipertrofica (10-20%) le patologie diagnosticate con più frequenza1,3,9.

Sono state descritte deformità toraciche nel 70-95% dei pazienti affetti da questa sindrome. Il pectus excavatum o pectus carinatum rappresentano le anomalie sternali associate1,3,9.
Il criptorchidismo appare in più della metà dei bambini con SN alla nascita (60-77%). Nelle bambine il menarca è solitamente ritardato, comparendo attorno ai 14 anni, anche se non coinvolge la fertilità. In entrambi i sessi la comparsa della pubertà è comunque ritardata1,3,9,11.
Le anomalie oculari e dell’udito sono relativamente frequenti in questi pazienti, con una incidenza rispettivamente del 55 e del 61%1,9.
In generale, i bambini affetti da SN manifestano un leggero ritardo motorio, che può essere dovuto alla ipotonia muscolare associata, soprattutto durante l’infanzia1. Il ritardo mentale si osserva nel 15-35% dei casi11,20. I problemi comportamentali che si possono osservare in questi pazienti consistono nel rallentamento psicomotorio, nell’irascibilità e testardaggine, nonché nella perturbazione del linguaggio consistente nel ripetere una frase o una parola appena udite. Sia le turbe percettive che l’iperattività sembrano ridursi verso i 9 anni, tuttavia il difetto di attenzione tende ad aumentare1,4,5,20,21. Per quanto riguarda il distretto orale, questi pazienti non hanno anomalie dentali patognomiche; solitamente presentano però carie e/o malattia parodontale in relazione alle cattive abitudini igienico-dietetiche, evidenziando così l’importanza della prevenzione19,22.
Diagnosi
Nella maggior parte dei casi la diagnosi si realizza clinicamente, nel 50% di essi è possibile confermarla geneticamente. Per quanto riguarda la diagnosi prenatale, si può considerare di essere in presenza di sindrome di SN nei feti polidramniotici, con versamento pleurale, edema e aumento del liquido amniotico pur mantenendo cariotipo normale. Si può realizzare anche una prova del DNA raccogliendo un campione del liquido amniotico o delle villosità corioniche1-3.
La diagnosi postnatale può realizzarsi clinicamente, basandosi su dei criteri maggiori e minori, descritti da Van Der Burgt nel 19941.
I criteri maggiori riferiscono:
- facies tipica;
- stenosi della valvola cardiaca polmonare;
- altezza sotto il terzo percentile;
- parente di primo grado affetto;
- ritardo mentale, criptorchidismo e displasia linfatica.
I criteri minori includono:
- facies suggestiva;
- altri difetti cardiaci;
- altezza sotto il decimo percentile;
- parente di primo grado con sospetta sindrome;
- una delle caratteristiche menzionate nei criteri maggiori.
La diagnosi della sindrome sarà definitiva se viene soddisfatto uno dei criteri indicati nella Tabella 3. La sindrome di Noonan è un’entità eterogenea che può essere riconosciuta clinicamente in relazione alla grande quantità di segni caratteristici presentati da questi pazienti2,19 (Tabella 2). La diagnosi differenziale di questa sindrome include: la sindrome di Turner, la sindrome di Leopard, la sindrome cardio-facio-cutanea e la sindrome di Costello 1,9,11,19.
Caso clinico
Paziente di 9 anni e 10 mesi di sesso maschile che giunge alla Clinica Universitaria della Catalunya. Al bambino veniva diagnosticata la sindrome di Noonan grazie ai criteri clinici a pochi mesi dalla nascita. Inoltre presentava: lieve deficit cognitivo, difficoltà motorie, siringomielia, quadro sindromico multiplo con stenosi dell’arco aortico destro, criptorchidismo e discreta ipoacusia transitoria.
Il paziente si avvaleva di aiuto psicologico ed era trattato con agente antipsicotico (Risperdal 0,05 mg) ogni 8 ore e con uno psicostimolante, Metilfenidato (Concerta 54 mg), una volta al giorno. Il motivo principale della prima visita era la presenza di carie. Durante questo incontro ci siamo avvalsi del parere del pediatra che consigliava una profilassi antibiotica in relazione alla patologia cardiaca presentata dal paziente.
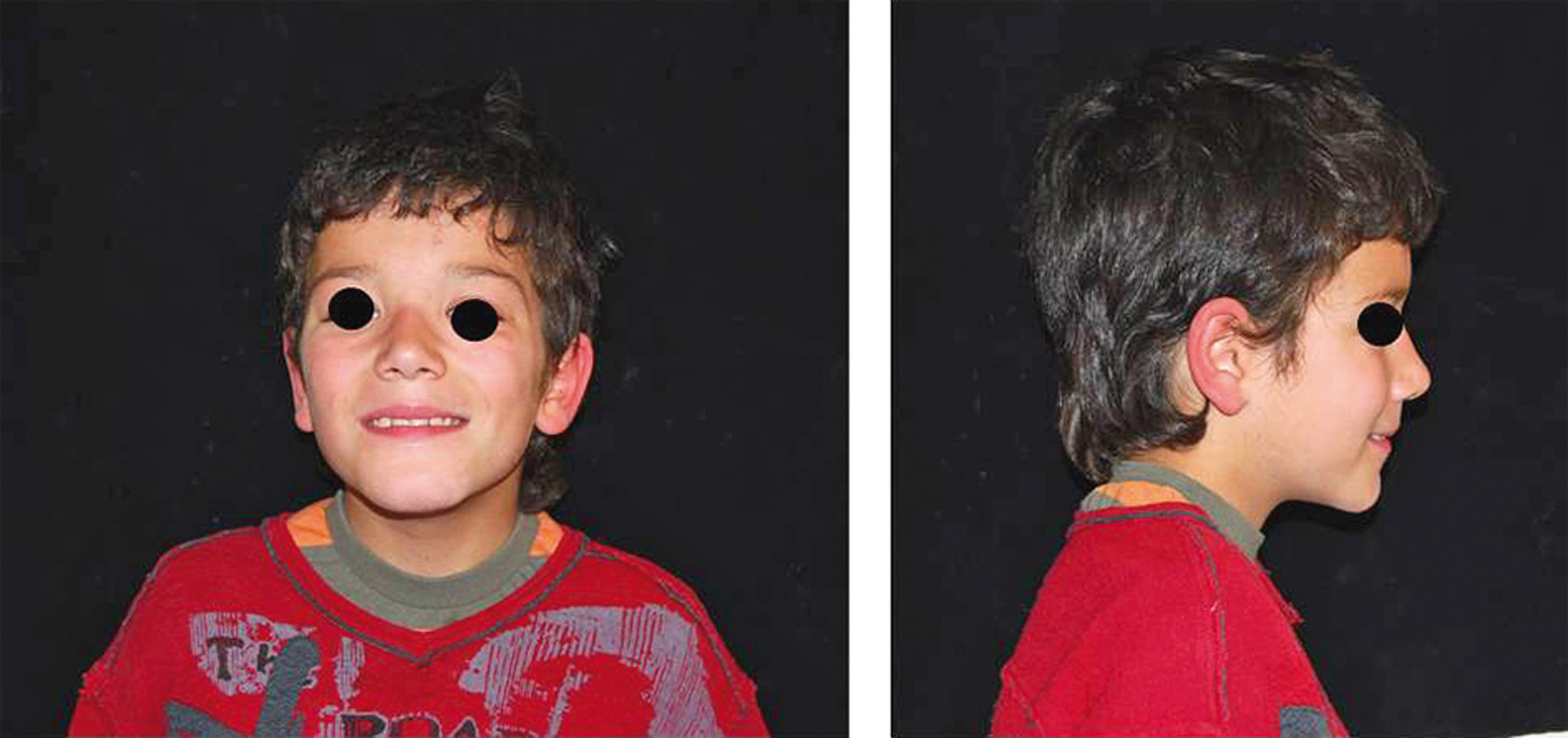
Si effettuava una visita clinica associata a esami radiografici. Durante l’esame obbiettivo si osservavano alcune caratteristiche frequenti associate al quadro sindromico: ipertelorismo, padiglione auricolare sottile inserito più basso, postero-rotazione auricolare, macchie di color caffelatte sulle orecchie e labbra sottili (Figura 1).
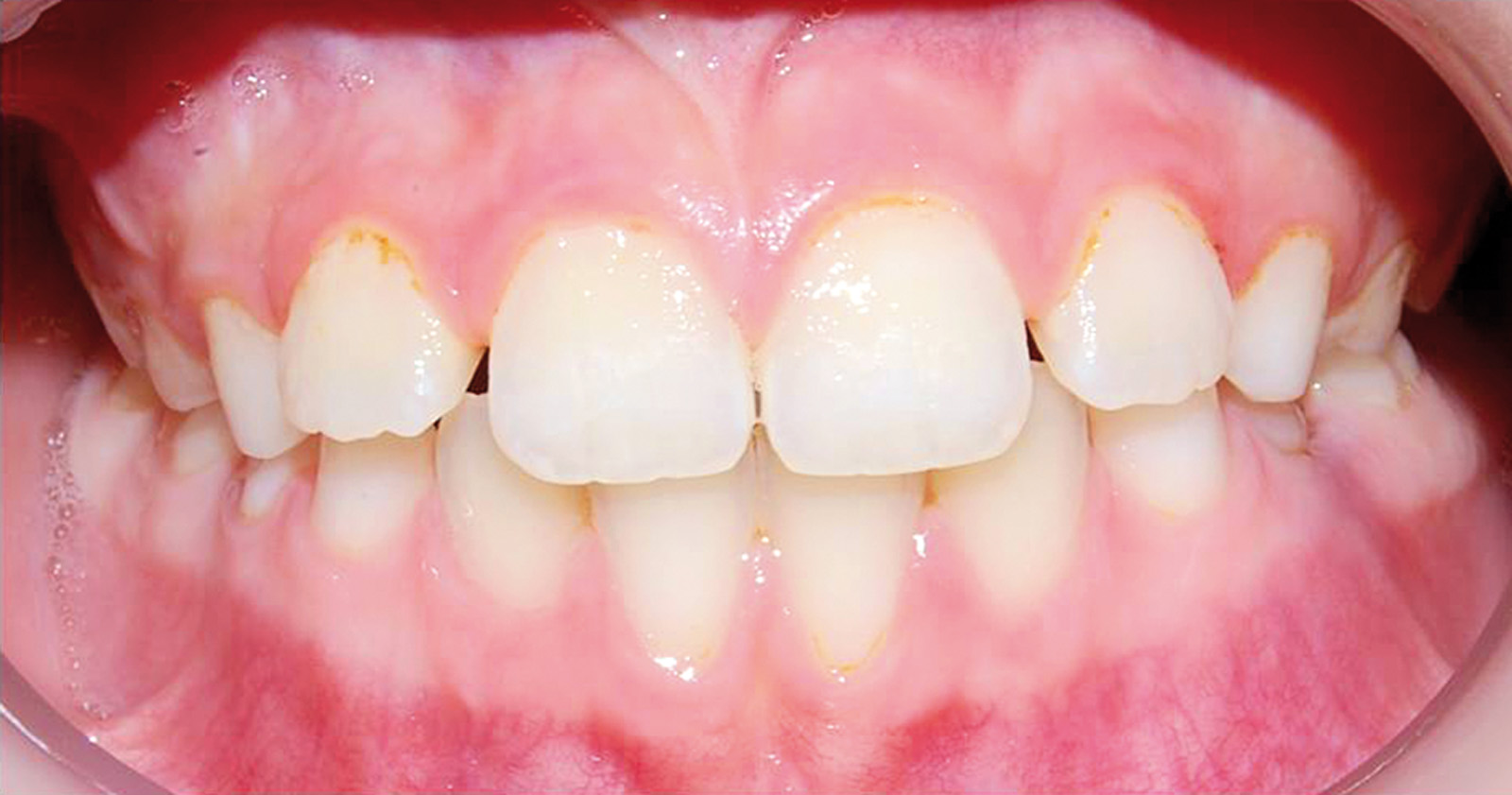
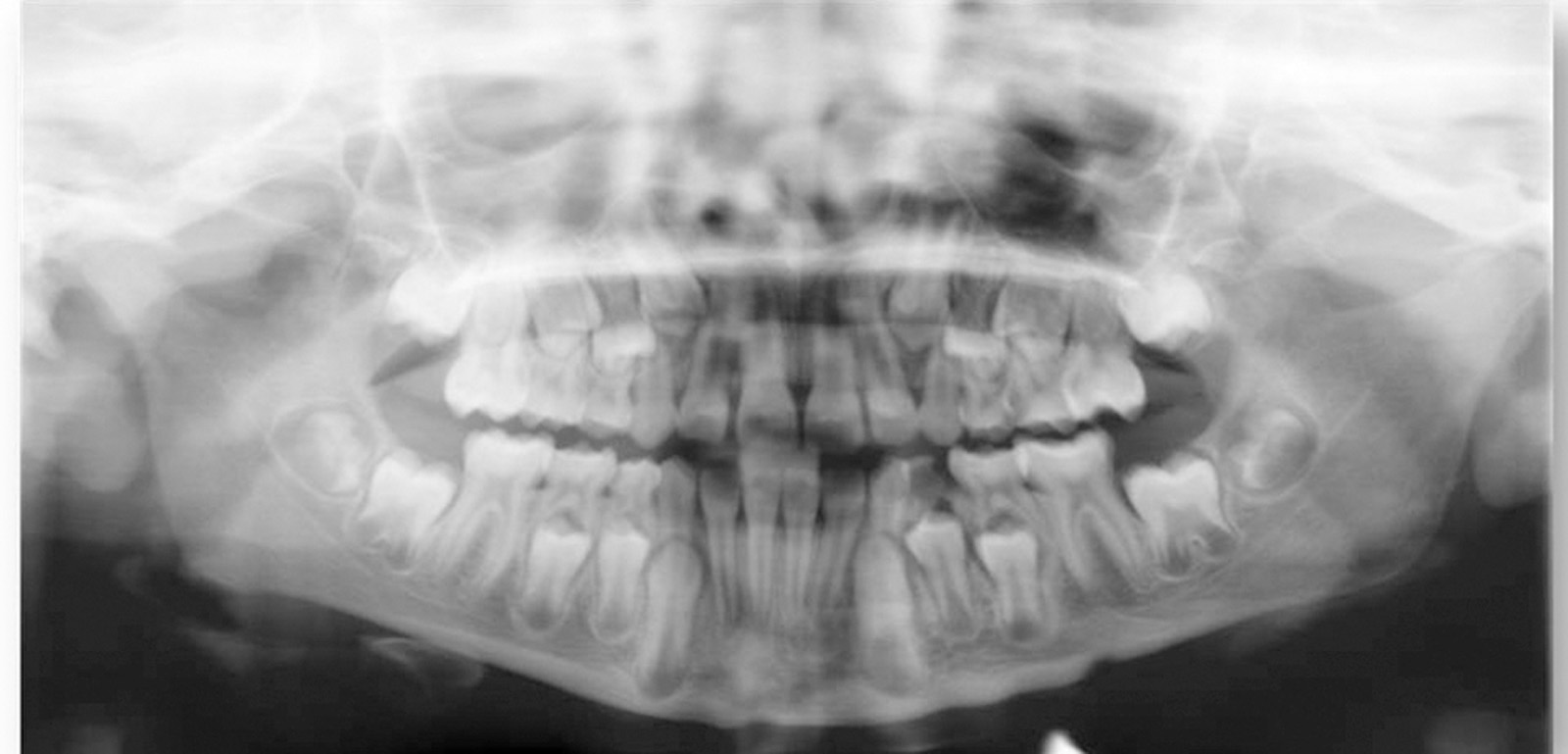
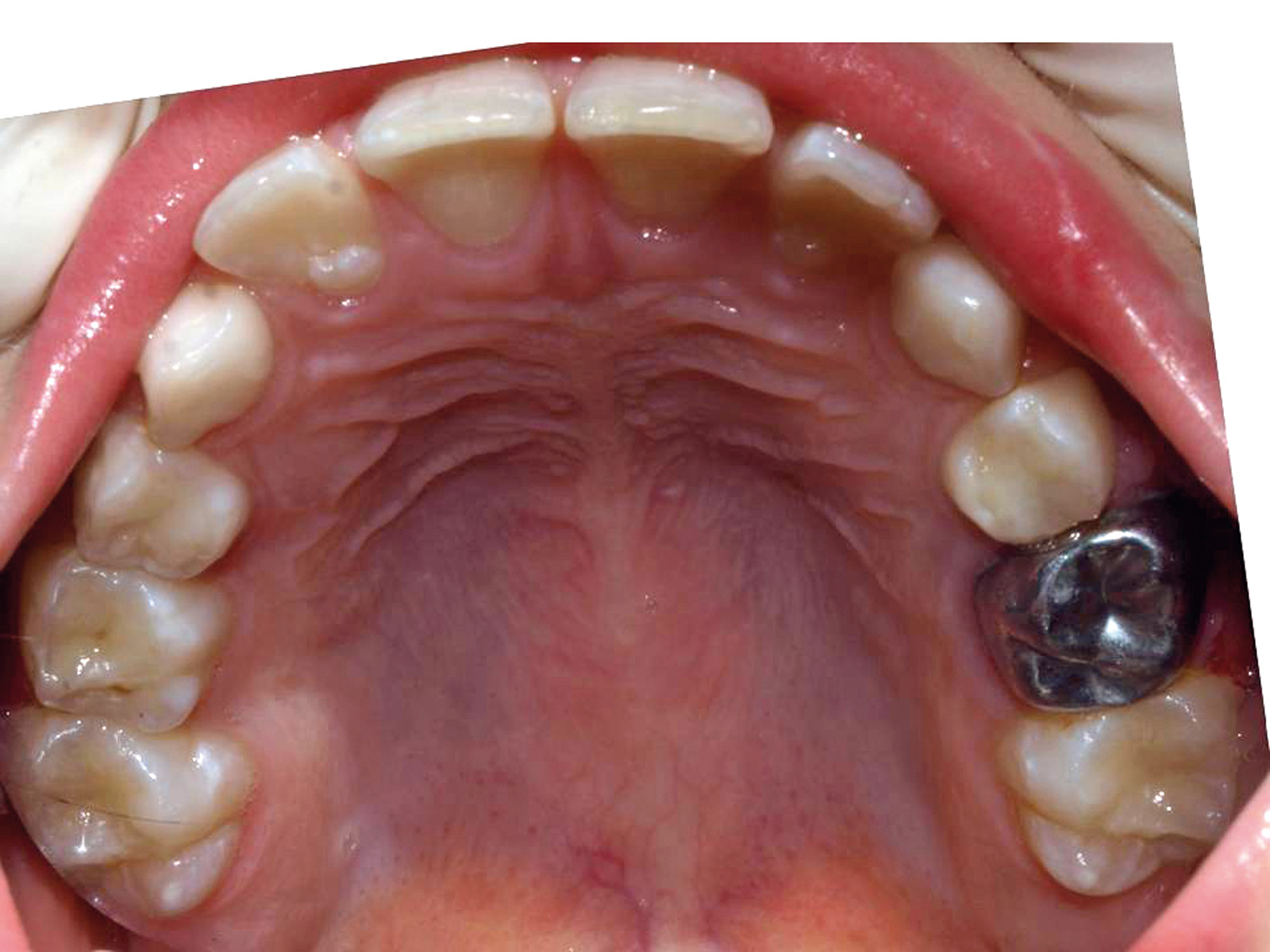
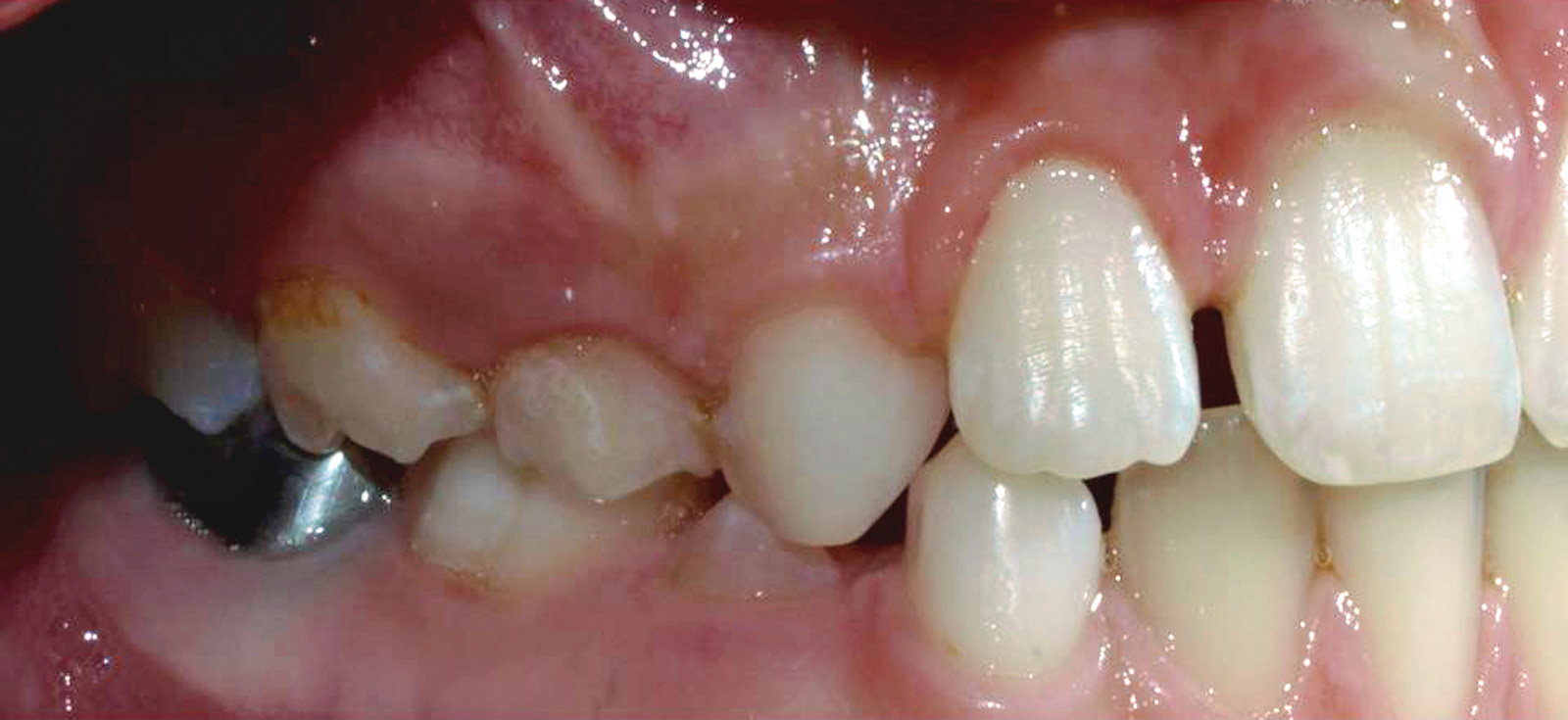
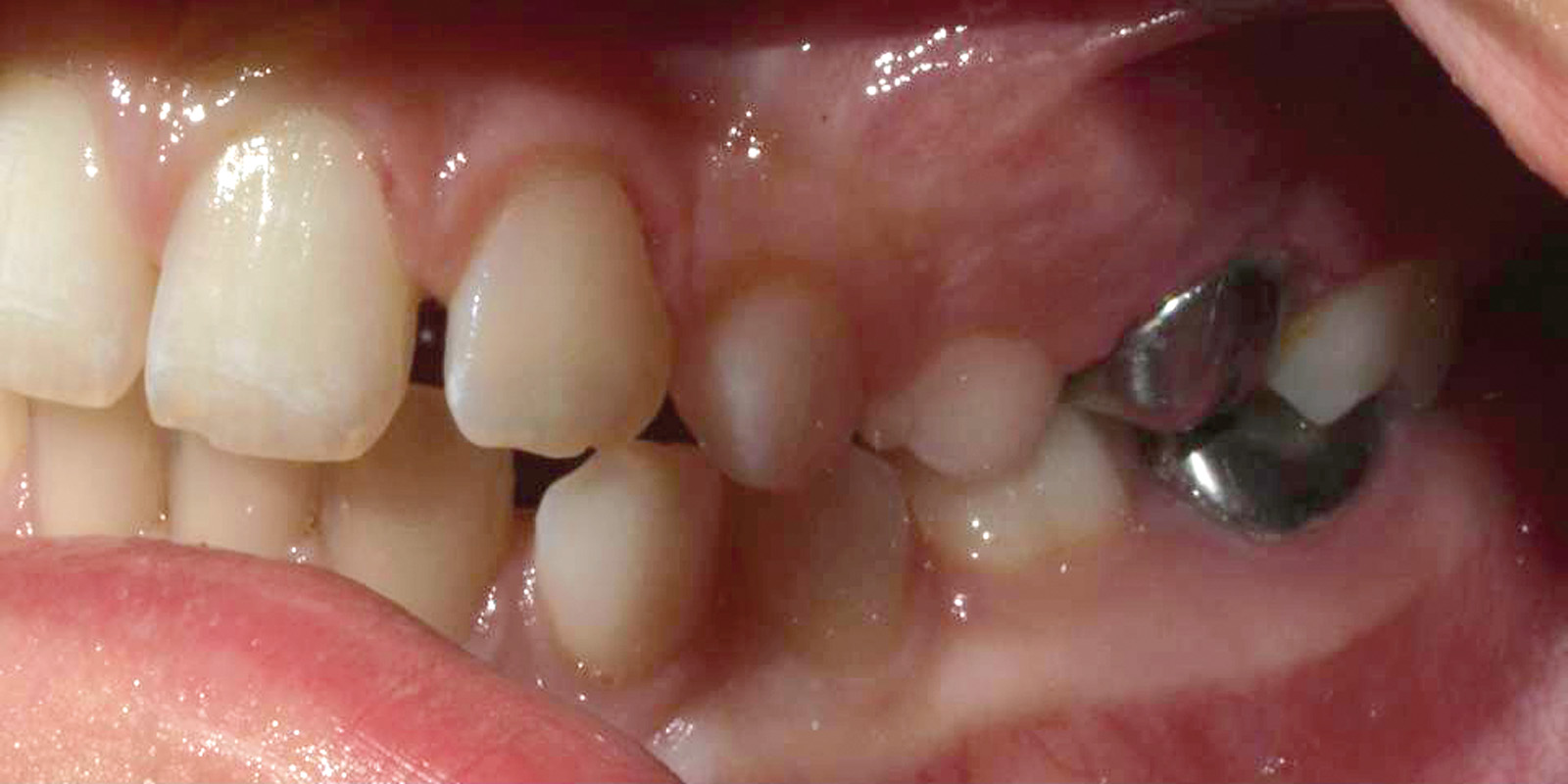
All’esame intraorale e al controllo radiografico si osservavano carie in 5.5, 5.4, 6.5, 8.4 e 7.4. e un’occlusione molare e canina in classe I. Il paziente presentava un’igiene orale insufficiente. I tessuti molli non evidenziavano anomalie, anche se era visibile una xerostomia provocata dai farmaci a cui il paziente era sottoposto, e la poca elasticità della muscolatura periorale. Durante la prima visita è stata notata la scarsa collaborazione e la bassa capacità di comprensione da parte del piccolo23 (Figure 2, 3a-c).
Trattamento conservativo
Dato il negativo approccio iniziale fornito dal paziente durante il primo incontro, si sono realizzate altre due visite al fine di sensibilizzarlo ulteriormente. In queste occasioni si faceva vedere lo strumentario comunemente utilizzato, così come quello che si sarebbe potuto usare nelle seguenti visite. La profilassi antibiotica decisa dal pediatra è stata: Amoxicillina 500 mg in sospensione orale ogni 12 ore.
Durante la quarta visita è stata praticata una seduta di igiene orale fornendo anche consigli igienico-dietetici tanto al bambino che ai genitori responsabili. Durante gli appuntamenti successivi sono stati realizzati i seguenti trattamenti restaurativi:
- otturazione di classe V di 5.5 e 5.4;
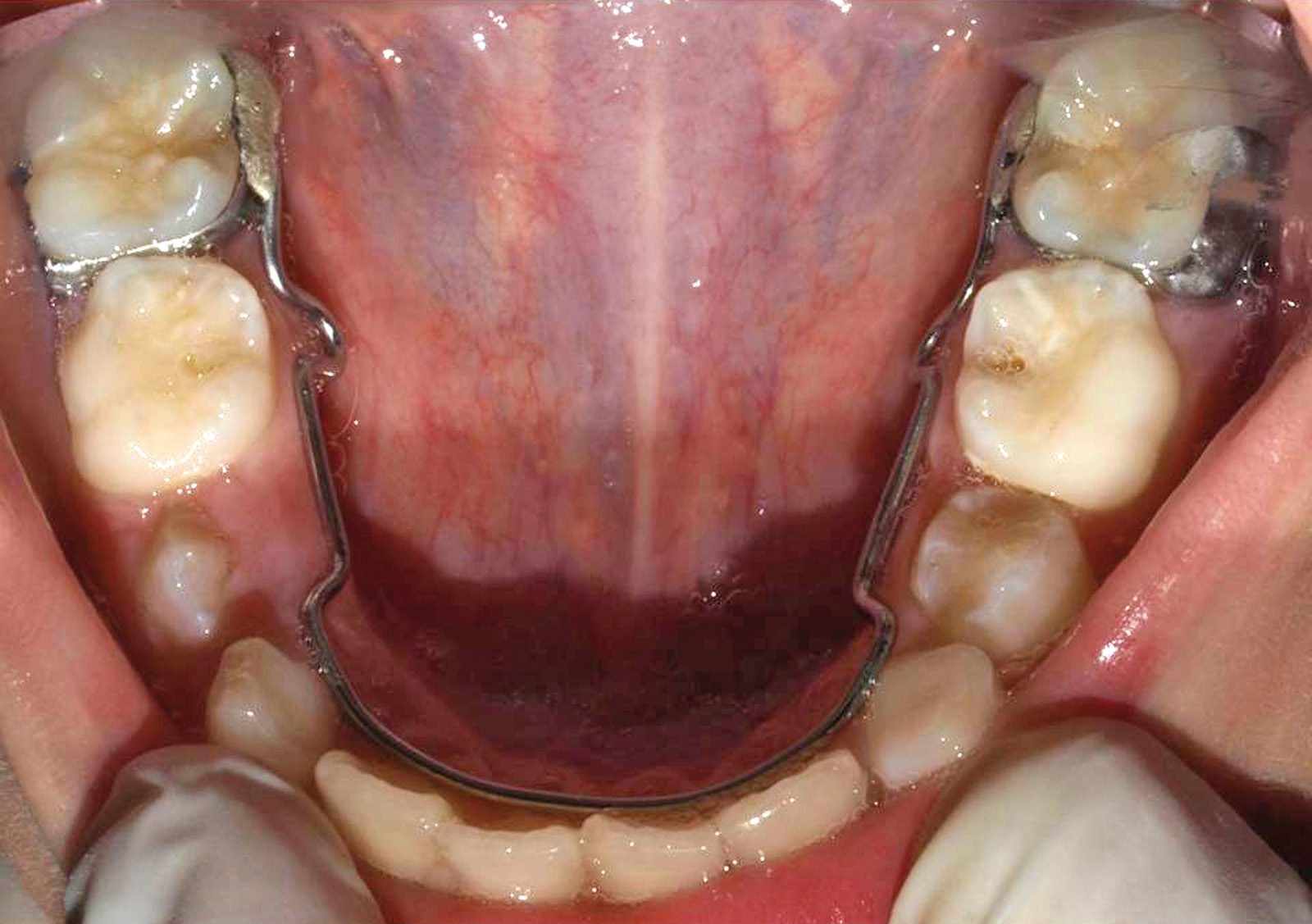
- prova di bande sui primi molari inferiori e impronta per la collocazione di un mantenitore di spazio bilaterale (arco linguale);
- pulpotomia con formocresol del 6.5;
- collocazione dell’arco linguale ed estrazione dell’8.4;
- corona metallica preformata del 6.5;
- estrazione del 7.4.
Anche se si è ricorsi a un rigoroso controllo del comportamento, il paziente si è mostrato incostante durante tutta la durata del trattamento, assumendo a volte anche un comportamento aggressivo.
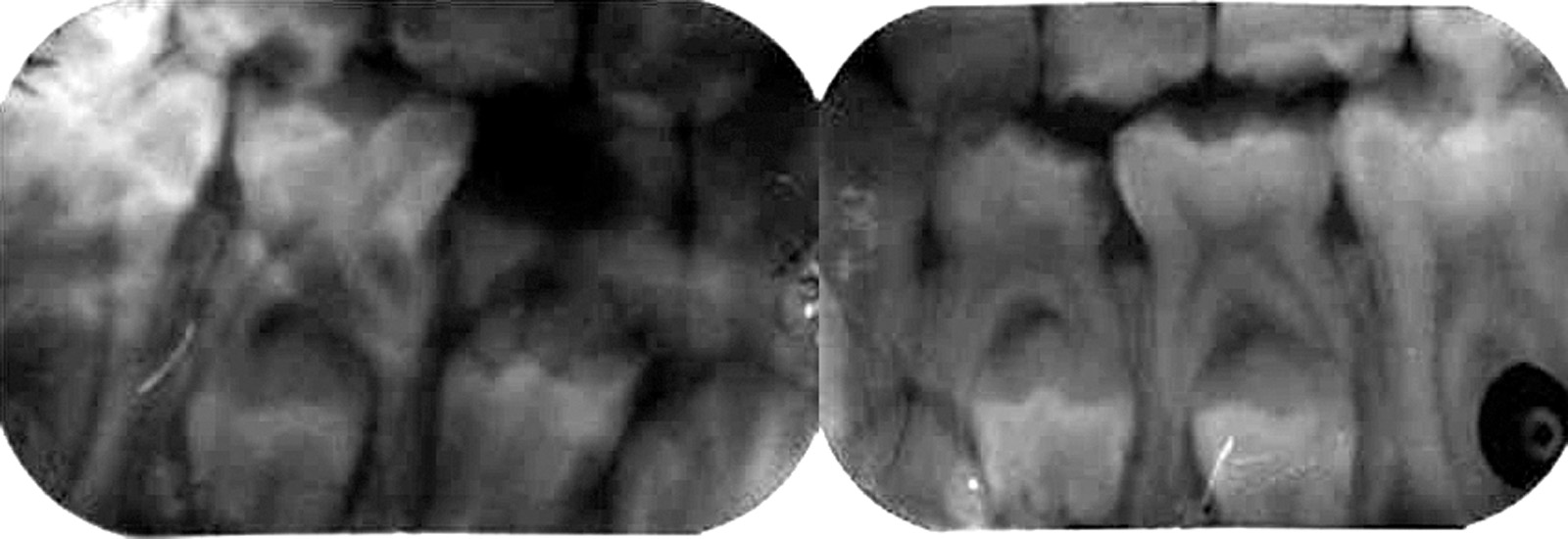
I tutori sono stati informati che sarebbero dovuti venire alle visite di controllo ogni 6 mesi, in realtà il bambino si è presentato all’appuntamento di controllo un anno dopo. In tale occasione si è riscontrata una carie occlusale del 7.5. L’età del paziente ci ha permesso di realizzare una radiografia periapicale, decidendo di controllare il bambino ogni 3 mesi essendo prossima la permuta del dente in questione (Figure 4a-4d).
Discussione
La sindrome di Noonan è un disordine dello sviluppo caratterizzato da dismorfismo facciale, bassa statura, difetti cardiaci e malformazioni scheletriche. L’eziologia continua a essere sconosciuta, anche se durante gli ultimi anni si sono scoperte mutazioni dei geni responsabili come: PTPN11, KRAS, SOS1, RAF1 e NF1.
La sindrome di Noonan è un disturbo genetico che ha un ampio spettro di manifestazioni.
La diagnosi può essere effettuata mediante criteri clinici maggiori e minori: si possono anche fare analisi ematologiche, genetiche, esami cardiaci, esami ormonali e valutazioni audiometriche.
Il trattamento sarà così personalizzato in base alla patologia specifica di ogni paziente e le aspettative dipenderanno dalla gravità dei sintomi.
Nel caso descritto è stato realizzato il trattamento conservativo che si è basato sullo sviluppo radicolare dei premolari e si è posizionato un mantenitore di spazio bilaterale inferiore.
Abbiamo fatto ricorso alle seguenti tecniche comportamentali: desensibilizzazione, tecnica del dire-mostrare-fare, imitazione e stimolo positivo. Tutte queste procedure non sono state efficaci in relazione al deficit cognitivo del paziente, quindi tutti i trattamenti si sono dovuti realizzare sotto restrizione fisica. Per tale motivo non siamo d’accordo con Ortega et al.19, che giungono alla conclusione che i bambini soggetti a SN non presentano nessun impedimento alla realizzazione dei trattamenti conservativi e, a nostro avviso, ogni caso deve essere affrontato in forma individuale e senza scartare l’opportunità di realizzare i trattamenti in anestesia generale.
In relazione alle necessità particolari di questi pazienti è necessario un trattamento multidisciplinare a tutti i livelli, fino al raggiungimento di uno sviluppo soddisfacente del bambino. Il pedodontista può essere il primo a fare una diagnosi precoce, per prevenire e risolvere i problemi odontoiatrici a breve e lungo termine.
Conclusioni
Si deve personalizzare ogni caso basandosi sui problemi specifici presentati da ogni piccolo paziente. Bisogna considerare la possibilità di un’anestesia generale per i pazienti con un comportamento estremo, quando con le semplici tecniche di gestione comportamentale non otteniamo risultati. ν
Corrispondenza/Correspondence
redaAna Isabel Lorente Rodriguez
Universitat Internacional de Catalunya - Facultad de Odontologia
Hospital General de Catalunya
Josep Trueta, s/n. 08190 St. Cugat del Vallès (Barcelona) - Espana
El síndrome de Noonan (SN) es un desorden congénito común originariamente llamado síndrome Ullrich-Turner. Fueron estos dos científicos, Ullrich en 1930 y Turner en 1938, quienes describieron un grupo de mujeres y hombres con un síndrome caracterizado por infantilismo sexual y un patrón de anomalías menores frecuentes, siendo la más común el cuello alado2,4. En 1883, Kobyslinsky describió el caso de un hombre joven con las características básicas del síndrome. Sin embargo, hasta 1963 no fue reconocido como entidad única por la pediatra y cardióloga Noonan en colaboración con Ehmke. Estos autores describieron una serie de pacientes con fascies inusuales y múltiples malformaciones, incluyendo defectos congénitos del corazón2,4,5. EtiologIa Su etiología sigue siendo poco clara, ya que se hereda de forma autosómica dominante con predominio de transmisión materna, aunque en el 60% de los casos aparece de forma esporádica1,2. En 2001, Tartaglia et al.6 descubrieron una serie de mutaciones en el gen PTPN11 del cromosoma 12q24.1 en el 50 % de los sujetos estudiados3,7,8. Este gen regula la producción de una proteína llamada SHP-2, la cuál es esencial en la transducción de diversas señales intracelulares y en el control del desarrollo de numerosos procesos, tales como el desarrollo del mesodermo y el desarrollo de la valvulogénesis semilunar cardíaca. Esta proteína se expresa en todo el cuerpo y juega un papel importante en la respuesta celular frente a los factores de crecimiento. Las mutaciones en PTPN11 en el SN dan lugar a un aumento de la función de la proteína SHP-2 y a una sobrerregulación de las rutas de transducción de señales celulares activadas por la proteína Ras y de las proteínas quinasas activadas por mitógenos (MAPK)9. La vía de señalización Ras-MAPK transduce señales de los factores de crecimiento y citoquinas, los cuáles participan en la transcripción de los genes involucrados en la proliferación celular y en la determinación del destino de las células. La vía Ras-MAPK es más conocida por su papel en la patogénesis del cáncer2,10-12. En 2005, Shubbert et al.13 descubrieron mutaciones en un segundo gen en pacientes que padecían SN. A este gen se le llamó KRAS. También causa un aumento en la función de la proteína SHP-2 y una sobrerregulación de la vía Ras-MAPK. Las mutaciones en este gen explican menos del 5% de los casos de SN11. A principios de 2007 se descubrió la mutación en el gen SOS1, que se presenta en el 15% de los casos, y codifica las proteínas implicadas en la vía Ras, causando la sobrerregulación de esta vía y un aumento de la señal de transducción. Los pacientes que presentan esta mutación presentan un aumento de la incidencia de rasgos ectodérmicos anormales (queratosis pilares facial), pelo rizado y estatura normal11. Existen mutaciones en un cuarto gen implicado (NF1), que causan pérdida de función de la proteína neurofibrina dando lugar a la neurofibromatosis tipo 1, enfermedad asociada a retraso mental, neurofibromas, manchas café con leche, nódulos metálicos en el iris y una tendencia a desarrollar tumores malignos11. Un quinto gen (RAF1) ha sido identificado como causante del SN, y fue relacionado con la aparición de cardiomiopatía hipertrófica4,14,15 (Tabla 1). Incidencia La incidencia de esta enfermedad es desconocida pero se sugiere que oscila de 1:1000 a 1:2500 nacidos vivos2, aunque Jamieson et al.7 describen una incidencia más baja, de 1:5000 nacidos vivos. Características clínicas Las características clínicas más importantes se describen en la Tabla 2. La dismorfia facial típica del síndrome de Noonan cambia a lo largo de la vida. En el recién nacido se observa excesiva piel en la parte de la nuca, como resultado de un higroma quístico fetal. A los tres o cuatro años, el cuerpo se muestra más fornido y el pecho más prominente. En esta edad, las deformidades del tórax a menudo empiezan a ser significativas. Durante la infancia, suelen tener la cabeza relativamente grande, ojos con hipertelorismo, ligeramente prominentes y redondos. El puente nasal suele ser plano, las orejas suelen estar posterorrotadas y con una inserción baja, y el cuello es corto con piel nucal redundante. Otros rasgos faciales típicos de esta etapa son: pliegues epicánticos, fisuras palpebrales oblicuas, paladar alto y arqueado y micrognatia9,11. En la adolescencia, la apariencia facial empieza a revelar rasgos más toscos, la cara tiene forma más triangular y el mentón aparece más alargado. En esta etapa, la ptosis comienza a hacerse evidente y el cuello es ligeramente más alargado. En la edad adulta el dismorfismo facial se atenúa pero el surco nasolabial es prominente. El cuello es alado y alargado, suelen tener pelo escaso y la línea de inserción anterior del cabello se sitúa en una posición más posterior, aparentando una frente más amplia. Los rasgos faciales, especialmente en la vejez, pueden ser más sutiles9,11. Una de las características clínicas más frecuentes, asociada al 70% de los casos, es la baja estatura. En el nacimiento, el peso y la talla son normales, aunque durante el primer año de vida el crecimiento se ve disminuido y la pubertad está retrasada dos años aproximadamente. En la edad adulta, la altura suele ser de 160 cm en los hombres y de 150 cm en las mujeres1,11, siendo desconocidos los factores causales. Se ha descrito una posible hipótesis asociada, que es la insuficiente secreción de la hormona del crecimiento (GH) o a una insuficiencia neurosecretora de ésta11,16, pero existe controversia sobre los efectos de la terapia con GH en relación al aumento de la altura en la edad adulta4,9,17,18. Otro signo clínico presente en el 80% de los pacientes afectados son los defectos congénitos del corazón, siendo la estenosis de la válvula pulmonar (50-62%) y la cardiomiopatía hipertrófica (10-20%) los que se detectan con más frecuencia1,3,9. Se han descrito deformidades de tórax en el 70-95% de los pacientes afectados por este síndrome. El pectus excavatum o pectus carianatum son las anomalías esternales asociadas1,3,9. La criptorquidia aparece en más de la mitad (60-77%) de los niños con SN en el momento del nacimiento. En las niñas la menarquia suele estar retrasada, apareciendo sobre los 14 años, aunque no afecta a la fertilidad. En ambos sexos el desarrollo puberal está retrasado1,3,9,11. Las anomalías oculares y de audición son relativamente frecuentes en estos pacientes, con una prevalencia del 55 y 61%, respectivamente1,9. En general, los niños con SN manifiestan un ligero retraso motor, el cuál puede ser debido a la hipotonía muscular asociada, sobre todo durante la infancia1. El retraso mental se observa en el 15-35% de los casos11,20. Los problemas de comportamiento que pueden observarse en estos pacientes son torpeza, nerviosismo, testarudez, ecolalia e irritabilidad. Los problemas de percepción pueden manifestarse junto con déficit de atención y/o hiperactividad. Tanto los problemas de percepción como la hiperactividad suelen atenuarse alrededor de los 9 años; en cambio, el déficit de atención tiende a aumentar con la edad1,4,5,20,21. Respecto a las características orales, estos pacientes no tienen anomalías dentales patognomónicas. Por ello, pueden presentar caries y/o enfermedad periodontal debido a malos hábitos higiénico-dietéticos, siendo importante hacer énfasis en las medidas preventivas19,22. Diagnóstico En la mayoría de los casos el diagnóstico se realiza clínicamente, pero en el 50% de los casos es posible confirmarlo genéticamente En referencia al diagnóstico prenatal, debe considerarse la posibilidad de padecer SN en los fetos con polihidramnios, derrame pleural, edema y aumento de líquido amniótico con cariotipo normal. También puede realizarse una prueba de ADN tomando muestras del líquido amniótico o vellosidades coriónicas1,2,3. El diagnóstico postnatal puede realizarse clínicamente, basándose en unos criterios mayores y menores, descritos por Van Der Burgt en 19941. Los criterios mayores refieren: Los criterios menores incluyen: El diagnóstico del síndrome será definitivo si cumple uno de los criterios que se indican en la Tabla 3. El SN es una entidad heterogénea, aunque puede ser reconocida clínicamente debido a la gran cantidad de rasgos atípicos que presentan estos pacientes2,19 (Tabla 2). El diagnóstico diferencial de este síndrome incluye: el síndrome de Turner, el síndrome de Leopard, el síndrome condrofaciocutáneo y el síndrome de Costello1,9,11,19. Caso clínico Paciente de 9 años y 10 meses de sexo masculino que acudió a la Clínica Universitaria Odontológica de la Universitat Internacional de Catalunya. El niño se diagnosticó de síndrome de Noonan mediante criterios clínicos a los pocos meses de nacer. Además presentaba: déficit cognitivo leve, trastorno motor moderado, siringomielia, síndrome polimalformativo, estenosis del arco aórtico derecho, criptorquidia y discreta hipoacusia transmisora. El paciente recibía ayuda psicológica y era tratado mediante el agente antipsicótico Risperidona (Risperdal® 0.05mg), cada 8 horas y un psicoestimulante, Metilfenidato (Concerta® 54mg), 1 vez al día. El motivo principal de consulta por el que acudió a la clínica fue: “Porque tiene caries”. Durante la primera visita, se realizó una interconsulta con la pediatra para pautar la profilaxis antibiótica debido a la patología cardiaca que padecía el paciente. Se le realizó una exploración clínica y radiográfica completa. Durante la exploración física se observaron algunas características frecuentemente asociadas al síndrome, tales como: hipertelorismo, hélix auricular fino, implantación baja, posterorrotación y manchas de color café con leche en las orejas y labios finos (Figura 1). En la exploración clínica intraoral y radiológica se observaron caries en 5.5, 5.4, 6.5, 8.4 y 7.4. y una oclusión molar y canina en clase I. Referente a los hábitos higiénicos, el paciente presentaba una higiene oral deficiente. Los tejidos blandos no presentaban anomalías, aunque presentaba xerostomía provocada por la medicación que recibía, y poca elasticidad de la musculatura perioral. Durante la primera cita se observó poca colaboración y comprensión por parte del paciente23 (Figura 2a, 3b y 3c). Tratamiento conservador Debido a la mala conducta que presentó en la primera visita, se realizaron dos visitas con la finalidad de desensibilizar al paciente. En estas sesiones se le mostró el instrumental más comúnmente empleado, así como el que se utilizaría en la siguiente visita. La pauta antibiótica profiláctica que pautó la pediatra fue: Amoxicilina 500 mg en suspensión oral cada 12 horas. En la cuarta visita se realizó la higiene oral y se dieron pautas higiénico-dietéticas tanto al niño como a los padres y cuidadores. En las citas posteriores se realizaron los siguientes tratamientos restauradores: A pesar de que se empleó un riguroso manejo de conducta, el comportamiento del paciente fue oscilante durante todo el tratamiento, e incluso en ocasiones presentó un comportamiento agresivo. Se informó a los tutores legales que se debía realizar una revisión a los 6 meses, pero este no acudió hasta los 12 meses. En este momento, se observó caries oclusal en 7.5. Por la edad del paciente se le realizó una radiografía periapical y se se decidió ir controlándolo cada 3 meses por su proximidad a la exfoliación (Figura 4a-4d). Discusión El síndrome de Noonan es un desorden del desarrollo caracterizado por dismorfia facial, estatura baja, defectos cardiacos y malformaciones esqueléticas. La etiología continua siendo desconocida, aunque en los últimos años se han descubiertos diversas mutaciones en genes responsables, tales como: PTPN11, KRAS, SOS1, RAF1 y NF1. El síndrome de Noonan es un desorden genético que tiene un amplio espectro de manifestaciones. Los pacientes son diagnosticados mediante criterios clínicos mayores y menores. También pueden ser diagnosticados mediante análisis hematológicos, análisis genéticos, exámenes cardiacos, alteraciones del desarrollo y evaluación auditiva. El tratamiento será individualizado, basándose en los patología específicas de cada paciente, y las expectativas serán dependientes de la severidad de los síntomas. En el caso descrito anteriormente, se realizó el tratamiento conservador necesario, y basándonos en el desarrollo radicular de los premolares, se colocó un mantenedor de espacio inferior bilateral. Se utilizaron las siguientes técnicas de manejo de conducta: desensibilización, técnica de decir-mostrar-hacer, imitación y refuerzo positivo. Todas ellas no fueron lo suficientemente exitosas debido al déficit cognitivo del paciente. Por ello, todos los tratamientos se tuvieron que realizar mediante restricción física. Por ello, no estamos de acuerdo con Ortega et al.19, quienes concluyen que los niños con SN no representan ningún impedimento a la hora de realizar un tratamiento odontológico exitoso. Por este motivo, cada caso debe valorarse de forma individualizada, y plantear la posibilidad de tratamiento bajo anestesia general. Debido a las necesidades especiales de estos pacientes, es necesario un tratamiento multidisciplinar a todos los niveles, para conseguir un correcto desarrollo del niño. El odontopediatra puede ser el primero en realizar un diagnóstico temprano, para prevenir y resolver los problemas odontológicos a corto y largo plazo. Conclusiones Se ha de individualizar cada caso basándonos en los problemas específicos que padece cada paciente. Valorar la anestesia general en pacientes con comportamiento extremos donde se observe que, pese a una correcta aplicación de las técnicas de manejo de conducta, no se progresa adecuadamente.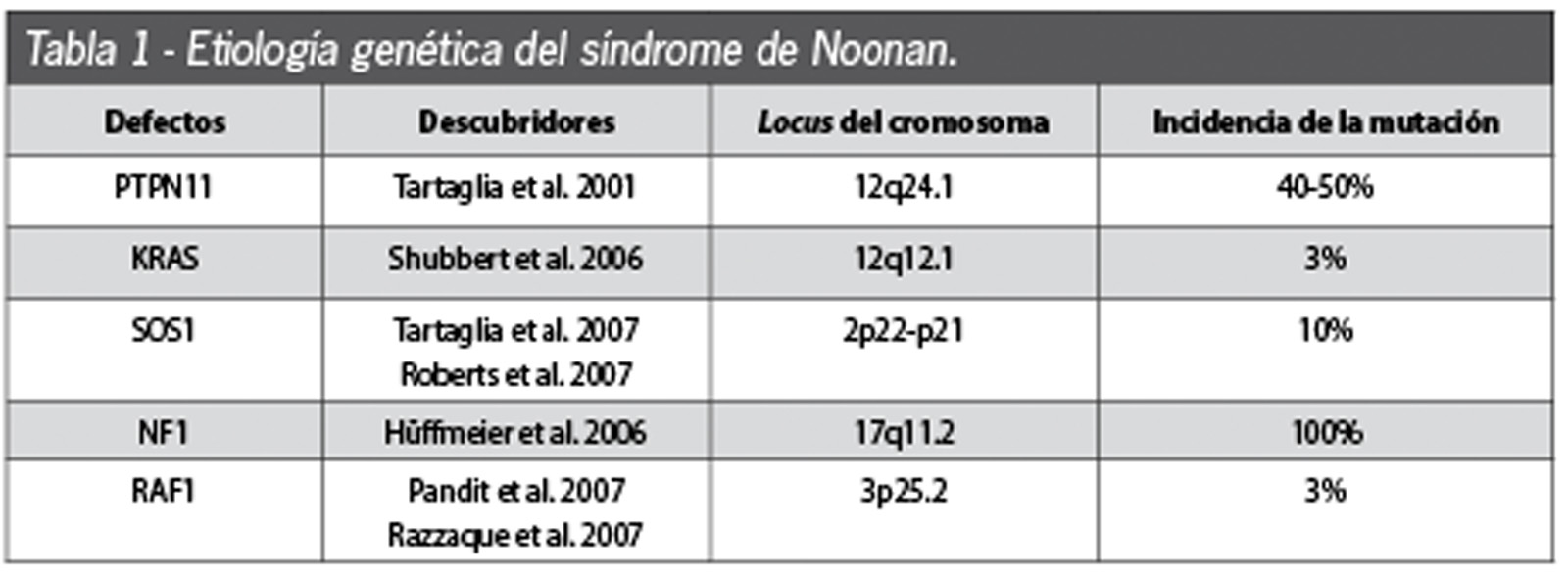








{kind=link}